Project
Objective: practice a wrap-up project that encompasses most of the workshop
Project - set-up
On your computer, in a folder with a meaningful name, create a new project using the project manager utility on the upper-right part of the rstudio window.
Check if you have all those libraries installed
library("tidyverse")
library("broom")
library("GEOquery")
theme_set(theme_bw(14)) # if you wish to get this theme by defaultAim
As you already experienced, working with GEO datasets can be a hassle. But it provides also a nice exercise as it requires to manipulate of a lot of tables (data.frame and/or matrix). Here, we will investigate the relationship between the expression of ENTPD5 and mir-182 as it was described by Pizzini et al.. Even if the data are already normalised and should be ready to use, you will see that reproducing the claimed results still requires an extensive amount of work.
Retrieve the GEO study
The GEO dataset of interest is GSE35834
- load the study using the
getGEOfunction
Warning
after NCBI moved its pages from http to https make sure that to have GEOquery version > 2.39
Solution
gse35834 <- getGEO("GSE35834", GSEMatrix = TRUE)## https://ftp.ncbi.nlm.nih.gov/geo/series/GSE35nnn/GSE35834/matrix/## OK## Found 2 file(s)## GSE35834-GPL15236_series_matrix.txt.gz## File stored at:## /var/folders/7x/14cplkhj0fn34yltb3c0j9bczm4jkt/T//RtmpmenFJR/GPL15236.soft## Warning in read.table(file = file, header = header, sep = sep, quote =
## quote, : not all columns named in 'colClasses' exist## GSE35834-GPL8786_series_matrix.txt.gz## File stored at:## /var/folders/7x/14cplkhj0fn34yltb3c0j9bczm4jkt/T//RtmpmenFJR/GPL8786.soft## Warning in read.table(file = file, header = header, sep = sep, quote =
## quote, : not all columns named in 'colClasses' existshow(gse35834)## $`GSE35834-GPL15236_series_matrix.txt.gz`
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 22486 features, 80 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: GSM875933 GSM875934 ... GSM876012 (80 total)
## varLabels: title geo_accession ... data_row_count (39 total)
## varMetadata: labelDescription
## featureData
## featureNames: 10000_at 10001_at ... 9_at (22486 total)
## fvarLabels: ID ENTREZ_GENE_ID Description SPOT_ID
## fvarMetadata: Column Description labelDescription
## experimentData: use 'experimentData(object)'
## Annotation: GPL15236
##
## $`GSE35834-GPL8786_series_matrix.txt.gz`
## ExpressionSet (storageMode: lockedEnvironment)
## assayData: 7815 features, 78 samples
## element names: exprs
## protocolData: none
## phenoData
## sampleNames: GSM875855 GSM875856 ... GSM875932 (78 total)
## varLabels: title geo_accession ... data_row_count (39 total)
## varMetadata: labelDescription
## featureData
## featureNames: 14q-0_st 14qI-1_st ... zma-miR408_st (7815 total)
## fvarLabels: ID miRNA_ID_LIST ... SEQUENCE (11 total)
## fvarMetadata: Column Description labelDescription
## experimentData: use 'experimentData(object)'
## Annotation: GPL8786- what kind of object is
gse35834?
Solution
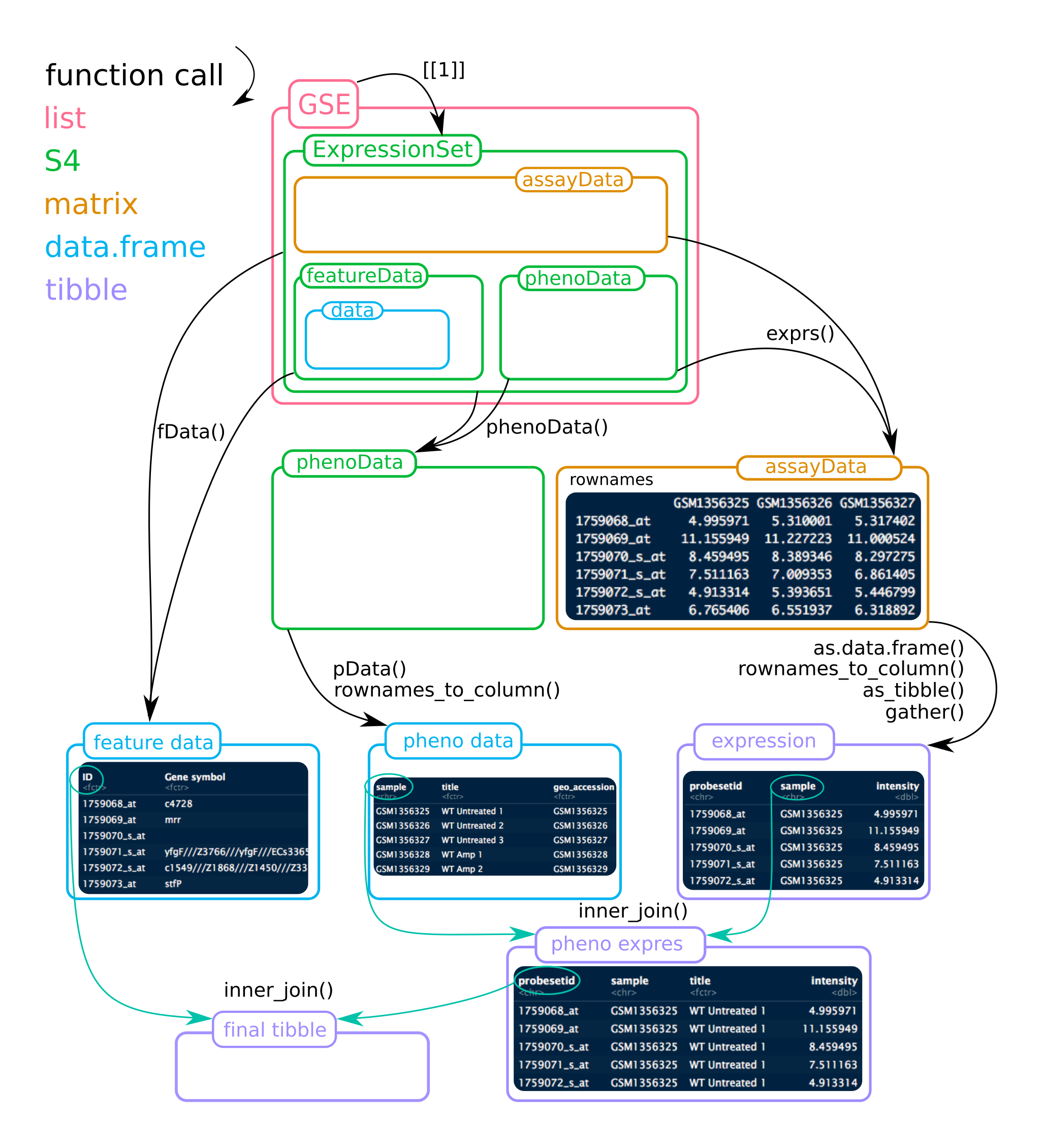
As shown in the Environment tab, it is a list composed of two elements. Each list is also a list with a special class ‘ExpressionSet’.
Tip
To help figuring out the ExpressionSet object, see the figure below. Mind that for this project, the list GSE contains 2 ExpressionSets!
- Two platforms were used in this study, which ones?
Solution
according to the GEO webpage:
- GPL15236 (HuEx-1_0-st Affymetrix Human Exon 1.0 ST Array)
- GPL8786 (miRNA-1_0 Affymetrix miRNA Array)
- How can you assign the mRNA or mir data to each element of
gse35834?
Solution
The function show() displays
- GPL15236
- GPL8786
Thus, gse35834[[1]] is mRNA (22486 probes) gse35834[[2]] is mir (7815 probes)
Explore the mRNA expression meta-data
You can use phenoData() to get informations on samples or pData() to retrieve them directly as a data.frame.
- Extract the mRNA meta-data as a
tibblewhich you will namerna_meta- rename
geo_accessiontosample - retain only the columns
source_name_ch1and starting with"charact"
- rename
Solution
rna_meta <- pData(gse35834[[1]]) %>%
as_tibble() %>%
select(sample = geo_accession,
source_name_ch1,
starts_with("charact"))Explore the mir expression meta-data
- Extract the mir meta-data as a
tibblewhich you will namemir_meta- rename
geo_accessiontosample - retain only the columns
source_name_ch1and all starting with “charact”
- rename
Solution
mir_meta <- pData(gse35834[[2]]) %>%
as_tibble() %>%
select(sample = geo_accession,
source_name_ch1,
starts_with("charact"))Join the meta-data
- Explore the two data frames with
View(rna_meta)andView(mir_meta). Are the samplesGSM*identical?
Solution
No, they aren’t. This is really annoying as the expression data contain only GSM ids.
We would like to somehow join both informations.
Knowing that both data frames have different sample columns, merge them to get the correspondence between RNA GSM* and mir GSM*. Save the result as rna_mir.
Note
If 2 data.frames that are joined (by specific columns) have identical names in their remaining columns, the default suffixes ‘.x’ and ‘.y’ are appended to the concerned column names from the first and second data frames respectively. However, you can make more friendly suffixes that match your actual data using the suffix = c(".x", ".y") option.
Solution
inner_join(rna_meta, mir_meta,
by = c("characteristics_ch1.1", "characteristics_ch1",
"source_name_ch1", "characteristics_ch1.2",
"characteristics_ch1.3", "characteristics_ch1.4",
"characteristics_ch1.5", "characteristics_ch1.6",
"characteristics_ch1.7", "characteristics_ch1.8"),
suffix = c("_rna", "_mir")) -> rna_mir## Warning: Column `characteristics_ch1.1` joining factors with different
## levels, coercing to character vector## Warning: Column `characteristics_ch1.3` joining factors with different
## levels, coercing to character vector## Warning: Column `characteristics_ch1.7` joining factors with different
## levels, coercing to character vectorGet RNA expression data for the ENTPD5 gene
Expression data can be accessed using exprs() which returns a matrix.
Warning
If you do not pipe the command to head, R would print ALL rows (or until it reaches max.print).
exprs(gse35834[[1]]) %>% head()- rows are probes and columns are sample ids in the form of
GSM*. - Probe ids by themselves are not meaningful, but
fData()provides features.
fData(gse35834[[1]]) %>% head()Again, we need to merge both informations to assign the expression data to the gene of interest.
- Find the common values that that allow us to join both data frames.
Solution
the probe ids are the common values
- The
rownamescontain the necessary informations. But as amatrixcontains, by definition, only a single data type (here numerical values), you will need to transform it to adata.frameand convert therownamesto a column usingrownames_to_column(var = "ID").
Save the result asrna_expression
Solution
exprs(gse35834[[1]]) %>%
as.data.frame() %>%
rownames_to_column(var = "ID") -> rna_expression- merge the expression data to the platform annotations (
fData(gse35834[[1]])). Save the result asrna_expression(Don’t worry:Ris always working on temporary objects and you won’t erase the object you are working on).
Note
Warnings about factors being coerced to characters can be ignored.
Solution
rna_expression %>%
inner_join(fData(gse35834[[1]])) -> rna_expression## Joining, by = "ID"## Warning: Column `ID` joining character vector and factor, coercing into
## character vector- Find the Entrez gene id for ENTPD5. Usually, the gene symbol is given in the annotation, but each GEO submission is a new discovery.
Solution
957, for Homo sapiens
- Filter
rna_expressionfor the gene of interest (ENTPD5) and tidy the samples:
A columnsamplefor allGSM*and a columnrna_expressioncontaining the expression values. Save the result asrna_expression_melt. At this point you should get atibbleof 80 values.
Solution
rna_expression %>%
filter(ENTREZ_GENE_ID == 957) %>%
gather(sample, rna_expression, starts_with("GSM")) -> rna_expression_melt- Add the meta-data and discard the columns
ID,SPOT_IDandsample_mir. Save the result asrna_expression_melt.
Solution
rna_expression_melt %>%
inner_join(rna_mir, by = c("sample" = "sample_rna")) %>%
select(-ID, -SPOT_ID, -sample_mir) -> rna_expression_melt## Warning: Column `sample`/`sample_rna` joining character vector and factor,
## coercing into character vectorGet mir expression data for miR-182
- Repeat the previous step but using
exprs(gse35834[[2]])for themir_expression. This time, the mir names are nicely provided byfData(gse35834[[2]])in the columnmiRNA_ID_LIST.
Solution
exprs(gse35834[[2]]) %>%
as.data.frame() %>%
rownames_to_column(var = "ID") %>%
# match expression data to platform annotation
inner_join(fData(gse35834[[2]])) %>%
gather(sample, mir_expression, starts_with("GSM")) %>% # melt patients
filter(miRNA_ID_LIST == "hsa-mir-182") -> mir_expression_melt## Joining, by = "ID"## Warning: Column `ID` joining character vector and factor, coercing into
## character vector- How many rows do you obtain? How many are expected?
Solution
78 samples for the mir experiment, so we would expect 78 but we are obtaining twice this number.
- Find out what happened, and plot the boxplot distribution of
expressionbyID
Solution
The mir array contains probes for both strands of mir:
- mature mir
- immature mir, named “*“, star.
Solution
mir_expression_melt %>%
ggplot(aes(x = ID, y = mir_expression)) +
geom_boxplot()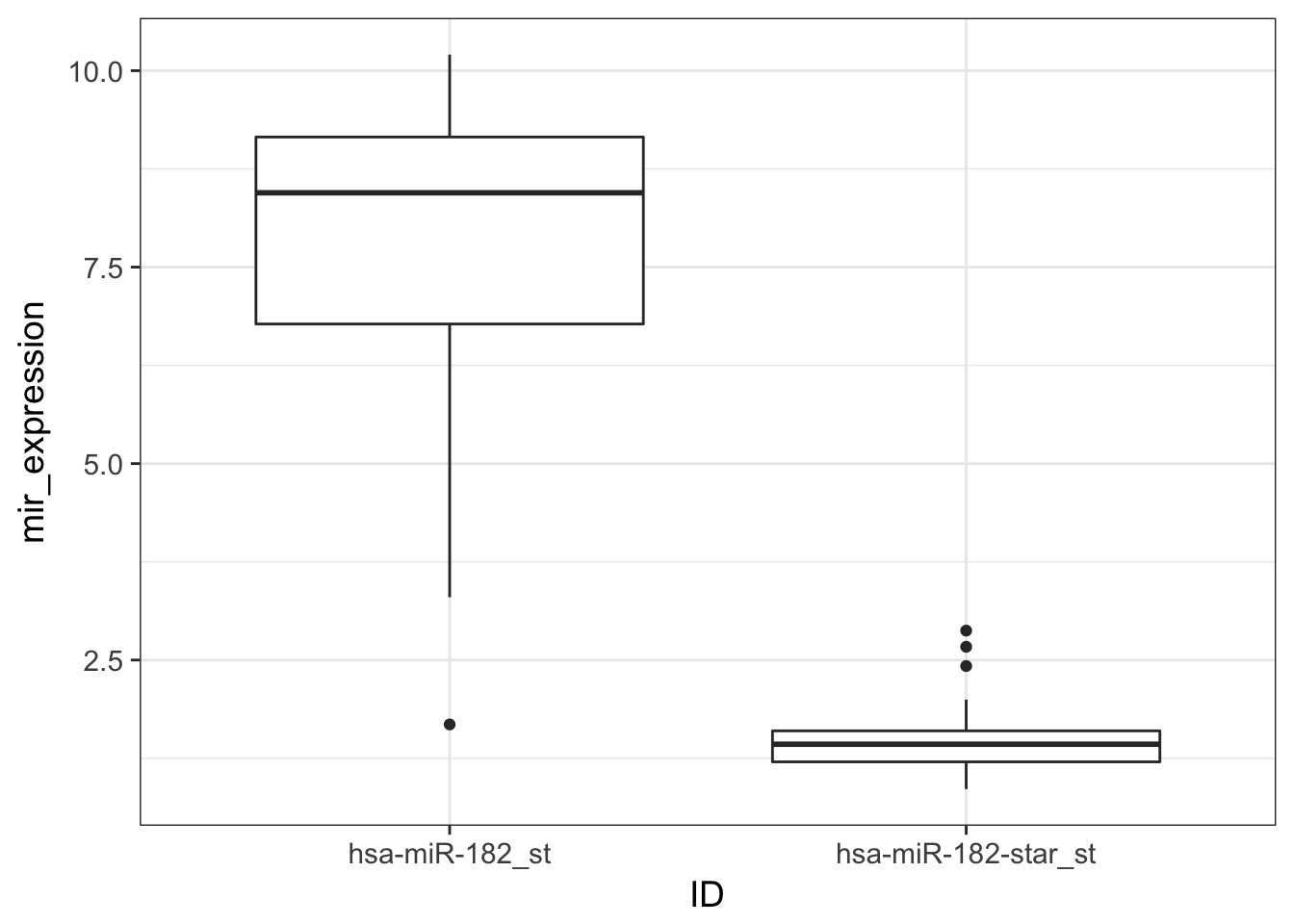
Solution
The immature mir, named star is indeed merely expressed
- Filter out the irrelevant IDs using
greplin thefilterfunction.
Hint
adding ! to a condition means NOT. Example filter(iris, !grepl("a", Species)): remove all Species containing the letter “a”.
Solution
mir_expression_melt %>%
filter(!grepl("star", ID)) -> mir_expression_melt- Add the meta-data, count the number of rows. Discard the column
sample_rnaafter joining.
Solution
mir_expression_melt %>%
inner_join(rna_mir, by = c("sample" = "sample_mir")) %>%
select(-sample_rna) -> mir_expression_melt## Warning: Column `sample`/`sample_mir` joining character vector and factor,
## coercing into character vectorSolution
77 rows, we lost GSM875854, which is not present in the meta-data nor the GSE description. Let it down
join both expression
Join rna_expression_melt and mir_expression_melt by their common columns EXCEPT sample. Save the result as expression.
Solution
expression <- inner_join(rna_expression_melt, mir_expression_melt,
by = c("source_name_ch1", "characteristics_ch1",
"characteristics_ch1.1", "characteristics_ch1.2",
"characteristics_ch1.3", "characteristics_ch1.4",
"characteristics_ch1.5", "characteristics_ch1.6",
"characteristics_ch1.7", "characteristics_ch1.8"))Examine gene expression according to meta data
- Plot the gene expression distribution by Gender. Is there any obvious difference?
Solution
expression %>%
ggplot(aes(y = rna_expression, x = characteristics_ch1)) +
geom_boxplot()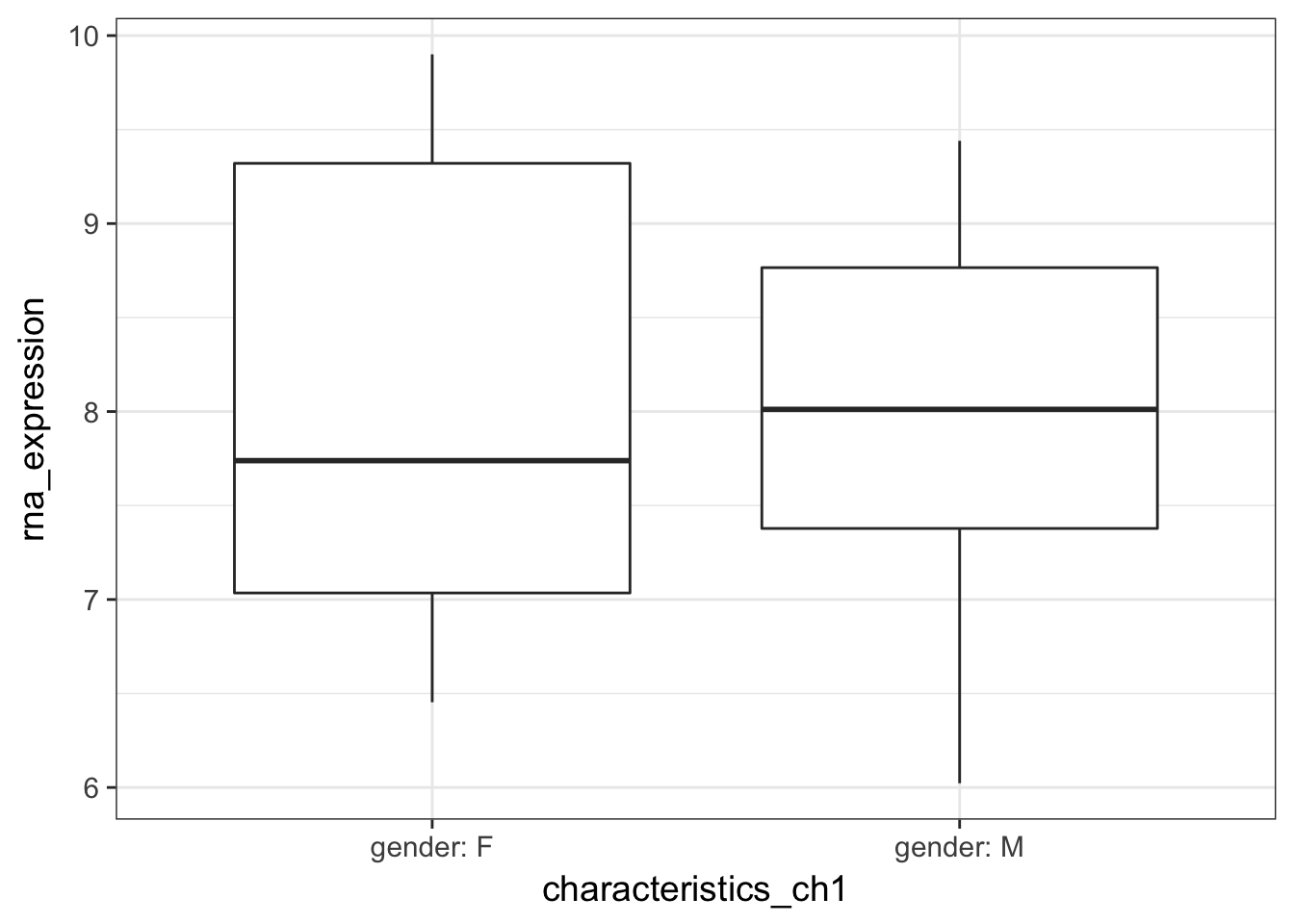
Solution
no relation to gender
- Plot gene AND mir expression distribution by Gender. Is there any obvious difference?
Hint
You will need to tidy by gathering rna and mir expression
Solution
expression %>%
gather(expression, value, ends_with("expression")) %>%
ggplot(aes(y = value, x = characteristics_ch1, fill = expression)) +
geom_boxplot()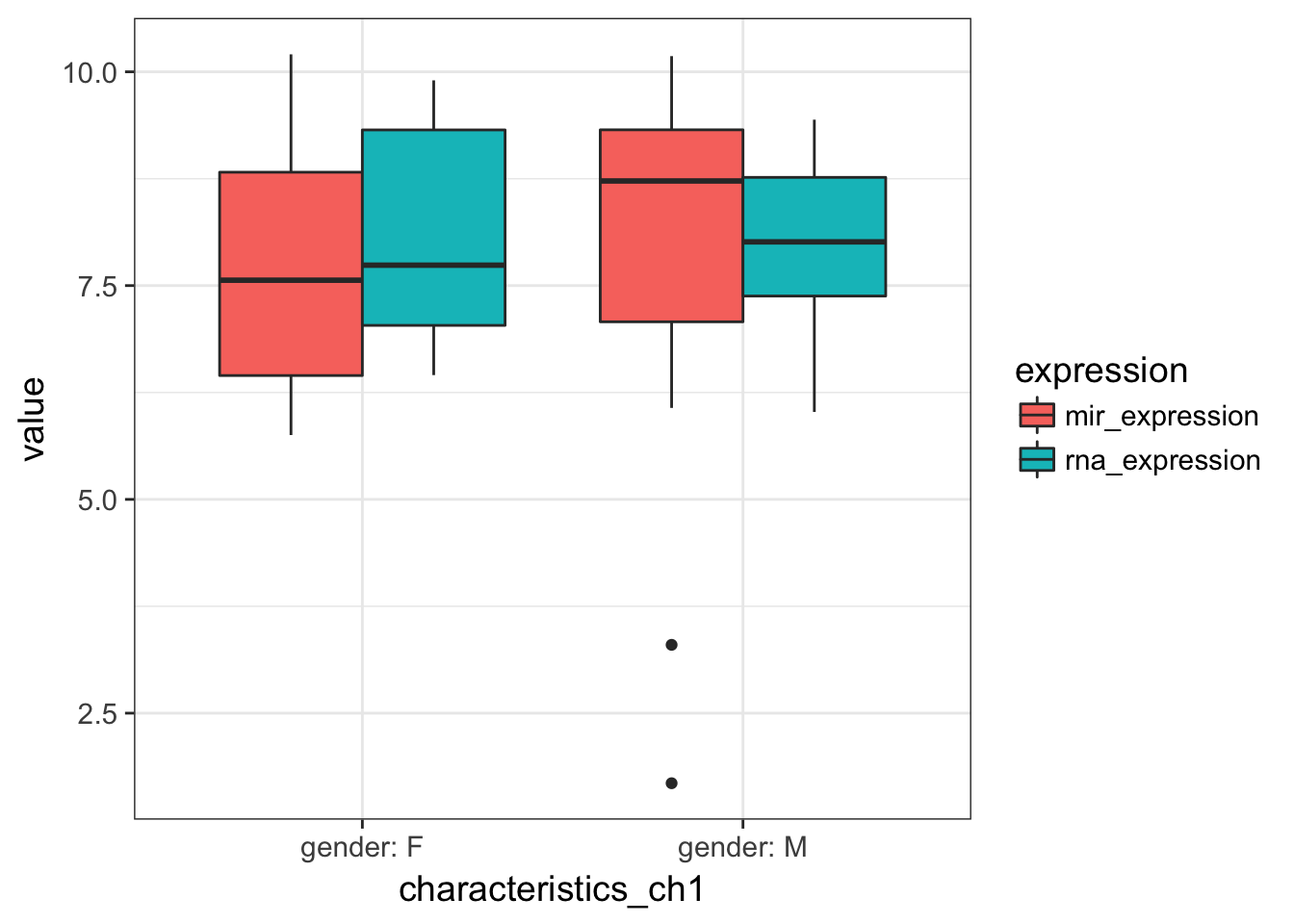
Solution
no relation to gender for both expressions
- Plot gene AND mir expression distributions by source (control / cancer). To make it easier, a quick hack is
separate(expression, source_name_ch1, c("source", "rest"), sep = 12)to getsourceas control / cancer. Is there any difference?
Solution
expression %>%
gather(expression, value, ends_with("expression")) %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
ggplot(aes(y = value, fill = source, x = expression)) +
geom_boxplot() + ggtitle("GSE35834")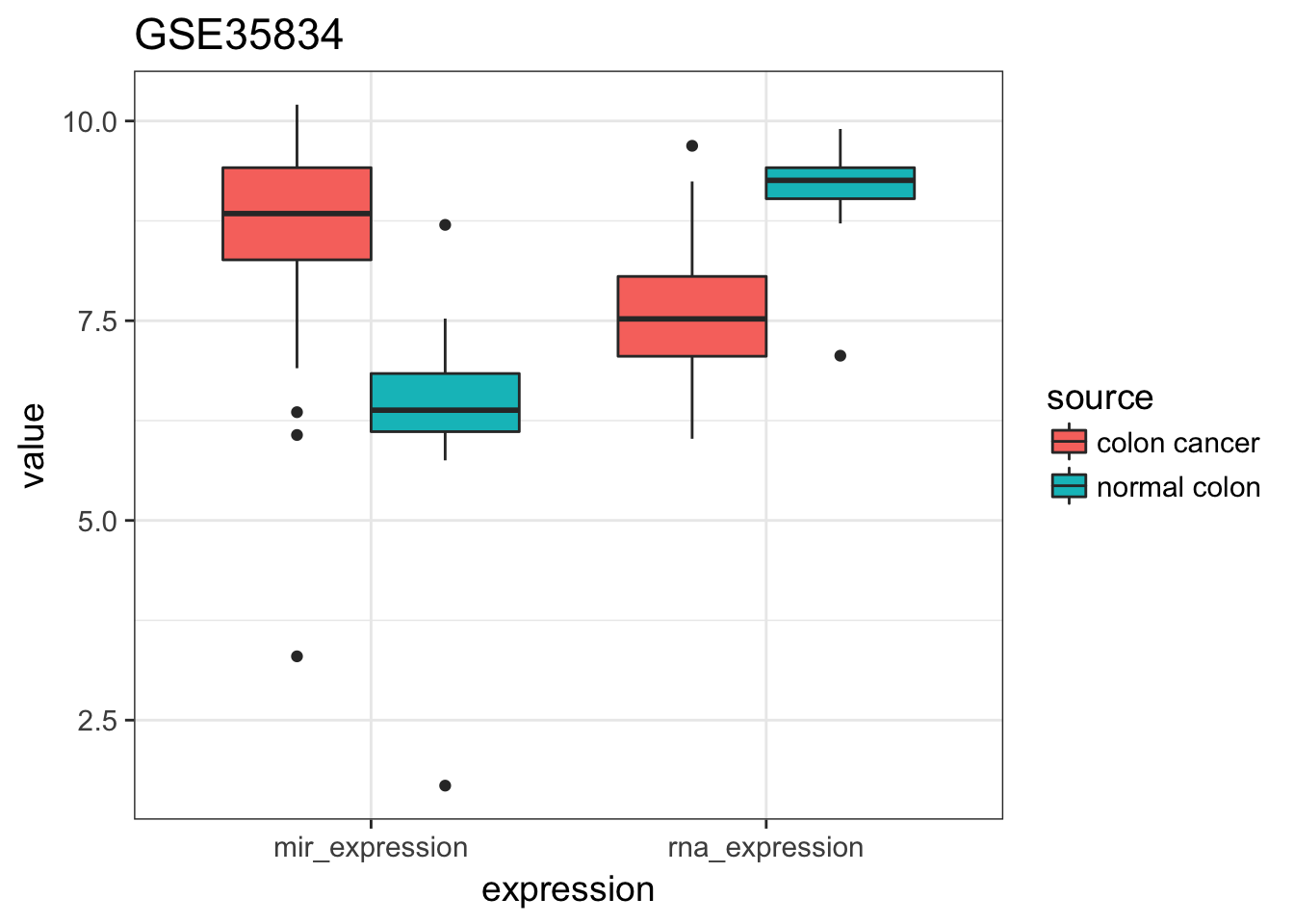
Solution
Like it is stated in the summary of the study, the expression of mir-182 seems indeed higher in cancer while the ENTPD5 expression seems lower.
- Do the same plot as in 3. but reorder the levels so that normal colon appears first. Display normal in “lightgreen” and cancer in “red” using
scale_fill_manual().
Solution
expression %>%
gather(expression, value, ends_with("expression")) %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
mutate(source = factor(source, levels = c("normal colon", "colon cancer"))) %>%
ggplot(aes(y = value, fill = source, x = expression)) +
geom_boxplot() +
scale_fill_manual(values = c("lightgreen", "red"))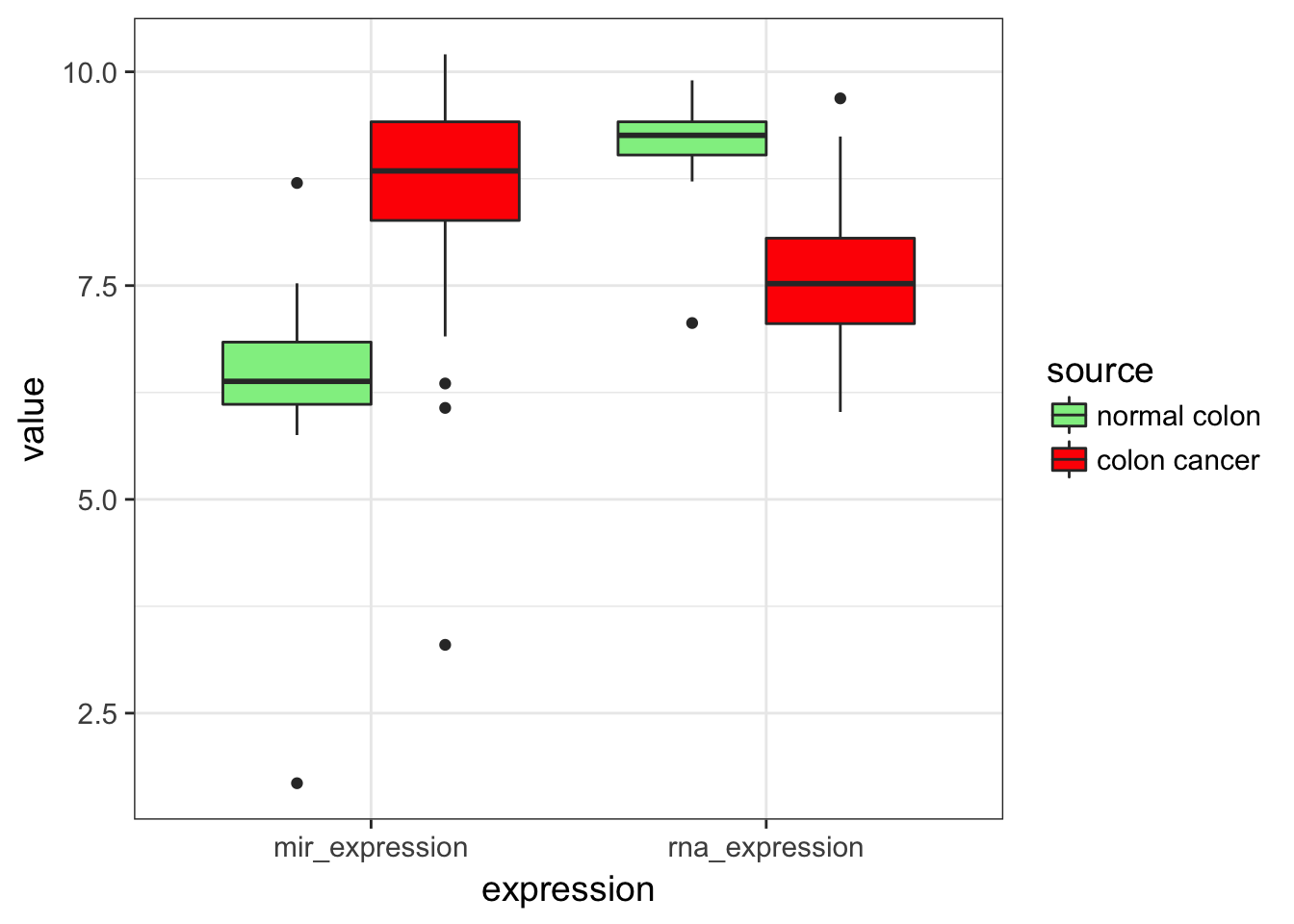
plot relation ENTPD5 ~ mir-182 as scatter-plot for all patients
- add a linear trend using
geom_smooth()for all data + per source
Solution
expression %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
ggplot(aes(x = mir_expression, y = rna_expression)) +
geom_point(aes(colour = source)) +
geom_smooth(method = "lm", se = FALSE) +
geom_smooth(aes(colour = source), method = "lm", se = FALSE) +
labs(y = "gene expression ENTPD5",
x = "hsa-mir-182 expression") +
ggtitle("GSE35834")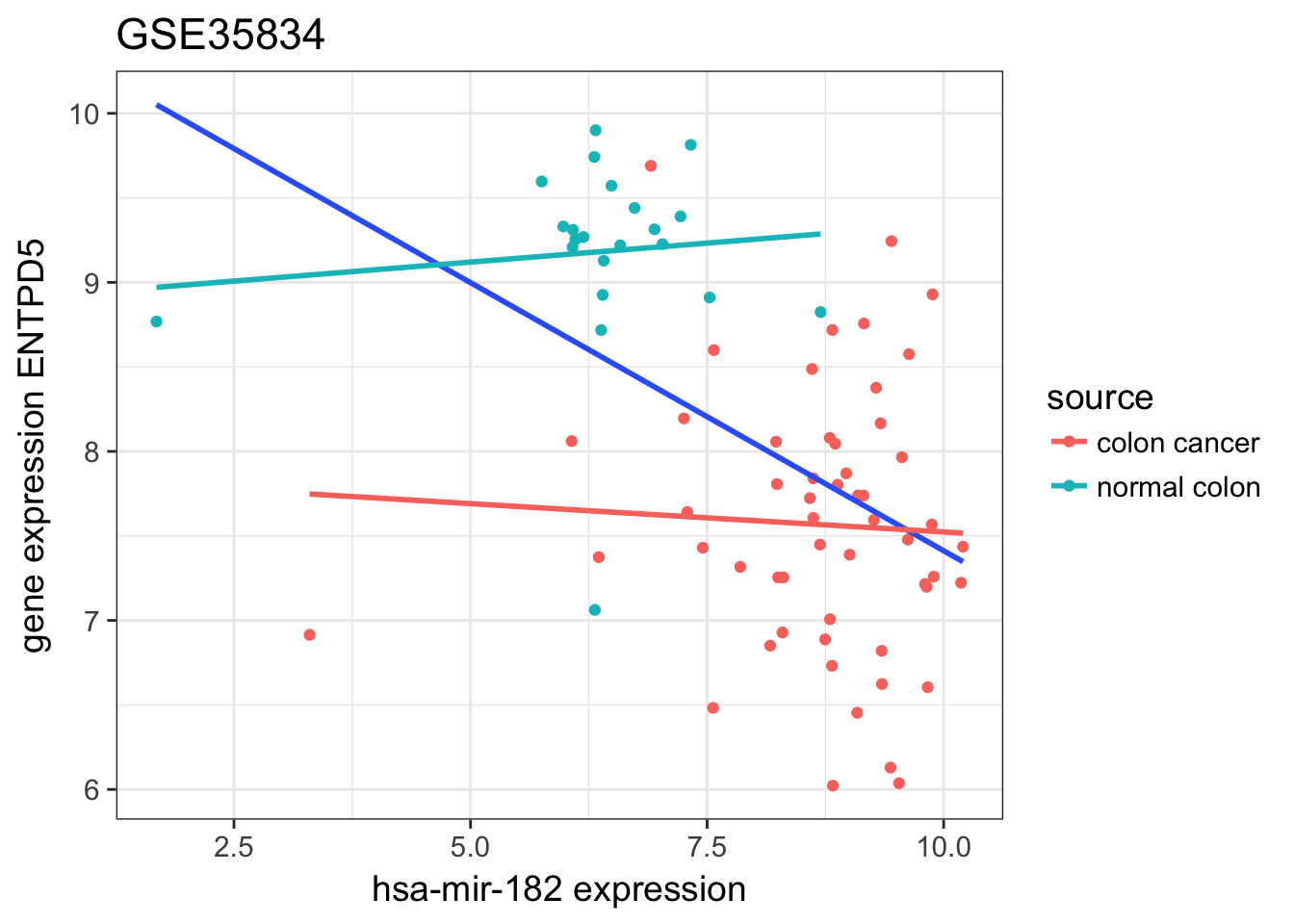
- does it support the claim of the study?
Solution
the two dot clouds between normal and cancer origin do split by
- high mir expression / low gene expression
- mild mir expression / high gene expression
but the trend is not so clear
linear models
- get the estimate from the linear trend for each source. linear models are outputted by
lm()as lists. Sincedata.frameare much easier to work with, David Robinson developedbroom.
Solution
library("broom")
expression %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
group_by(source) %>%
do(tidy(lm(rna_expression ~ mir_expression, data = .))) %>%
filter(term != "(Intercept)")## # A tibble: 2 x 6
## # : Groups: source [2]
## source term estimate std.error statistic p.value
## <chr> <chr> <dbl> <dbl> <dbl> <dbl>
## 1 colon cancer mir_expression -0.03354124 0.09217281 -0.3638951 0.7174117
## 2 normal colon mir_expression 0.04496954 0.10042656 0.4477853 0.6588934Solution
# the new purrr way
expression %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
group_by(source) %>%
nest() %>%
mutate(model = map(data, ~ lm(rna_expression ~ mir_expression, data = .x)),
tidy = map(model, tidy -> expression_lm
expression_lm %>%
select(source, tidy) %>%
unnest(tidy) %>%
filter(term != "(Intercept)")The estimate of the intercept is not meaningful thus it is filtered out. One can easily see that the slope is not significant when data are slipped by source.
- Perform the linear regression and tidy the results for all data, is it significant?
Solution
expression %>%
do(tidy(lm(rna_expression ~ mir_expression, data = .))) %>%
filter(term != "(Intercept)")## term estimate std.error statistic p.value
## 1 mir_expression -0.3172285 0.06592259 -4.812137 7.545623e-06Solution
with a pvalue of 7.54e-6, the negative is highly significant
- replace
tidybyglanceto extract the \(r^2\). Is this value satisfactory?
Solution
expression %>%
do(glance(lm(rna_expression ~ mir_expression, data = .)))## r.squared adj.r.squared sigma statistic p.value df logLik
## 1 0.2359153 0.2257275 0.9136575 23.15666 7.545623e-06 2 -101.292
## AIC BIC deviance df.residual
## 1 208.584 215.6154 62.60775 75Solution
with a r^2 of 0.236, i.e only 23.6% of the variance explained, a linear fit sounds bad due to outliers
Perform a linear model for the expression of ENTPD5 and ALL mirs
- Count how many
hsa-mir, which are not star, are present on the array GPL8786
Solution
fData(gse35834[[2]]) %>%
filter(grepl("^hsa", ID)) %>%
filter(!grepl("star", ID)) %>%
nrow()## [1] 677- Retrieve the expression values for the 677 human mir like you did before. Same procedure, except that you don’t filter for mir-182. Save as
all_mir_rna_expression
Solution
exprs(gse35834[[2]]) %>%
as.data.frame() %>%
rownames_to_column(var = "ID") %>%
filter(grepl("^hsa", ID)) %>%
# match expression data to platform annotation
gather(sample, mir_expression, starts_with("GSM")) %>%
filter(!grepl("star", ID)) %>%
inner_join(fData(gse35834[[2]])) %>%
inner_join(rna_mir, by = c("sample" = "sample_mir")) %>%
select(-sample_rna) %>%
inner_join(rna_expression_melt,
by = c("source_name_ch1", "characteristics_ch1",
"characteristics_ch1.1", "characteristics_ch1.2",
"characteristics_ch1.3", "characteristics_ch1.4",
"characteristics_ch1.5", "characteristics_ch1.6",
"characteristics_ch1.7", "characteristics_ch1.8"),
suffix = c("_mir", "_rna")) -> all_mir_rna_expression ## Joining, by = "ID"## Warning: Column `ID` joining character vector and factor, coercing into
## character vector## Warning: Column `sample`/`sample_mir` joining character vector and factor,
## coercing into character vector- Perform the 677 linear models, tidy the results and arrange by the
adj.r.squared
Solution
all_mir_rna_expression %>%
group_by(ID) %>%
do(glance(lm(rna_expression ~ mir_expression, data = .))) %>%
ungroup() %>%
arrange(desc(adj.r.squared))## # A tibble: 677 x 12
## ID r.squared adj.r.squared sigma statistic
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 hsa-miR-378_st 0.5337446 0.5275279 0.7137146 85.85605
## 2 hsa-miR-422a_st 0.3880594 0.3799002 0.8176497 47.56092
## 3 hsa-miR-215_st 0.3649550 0.3564878 0.8329423 43.10187
## 4 hsa-miR-145_st 0.3239984 0.3149851 0.8593825 35.94649
## 5 hsa-miR-183_st 0.3066374 0.2973925 0.8703479 33.16851
## 6 hsa-miR-17_st 0.2964648 0.2870843 0.8767093 31.60447
## 7 hsa-miR-106a_st 0.2875369 0.2780374 0.8822545 30.26861
## 8 hsa-miR-140-3p_st 0.2812168 0.2716330 0.8861589 29.34301
## 9 hsa-miR-138_st 0.2693901 0.2596486 0.8934195 27.65396
## 10 hsa-miR-139-5p_st 0.2689871 0.2592402 0.8936659 27.59736
## # ... with 667 more rows, and 7 more variables: p.value <dbl>, df <int>,
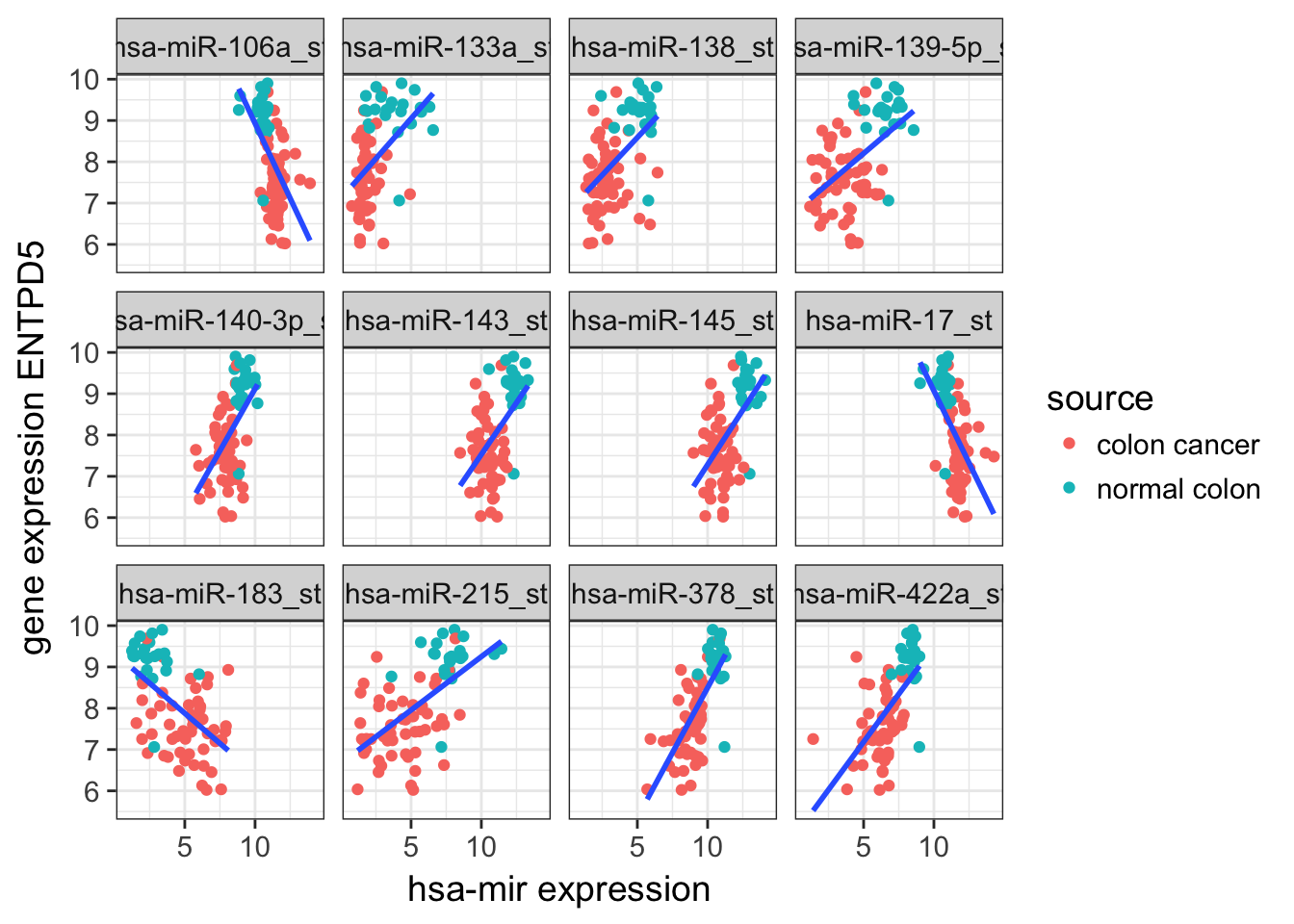
## # logLik <dbl>, AIC <dbl>, BIC <dbl>, deviance <dbl>, df.residual <int>- Get the top 12 mir and plot the scatter plot
Solution
top12_mir <- all_mir_rna_expression %>%
group_by(ID) %>%
do(glance(lm(rna_expression ~ mir_expression, data = .))) %>%
ungroup() %>%
arrange(desc(adj.r.squared)) %>%
head(12) %>%
.$ID
all_mir_rna_expression %>%
filter(ID %in% top12_mir) %>%
separate(source_name_ch1, c("source", "rest"), sep = 12) %>%
ggplot(aes(x = mir_expression, y = rna_expression)) +
geom_point(aes(colour = source)) +
geom_smooth(method = "lm", se = FALSE) +
facet_wrap(~ ID, ncol = 4) +
labs(y = "gene expression ENTPD5",
x = "hsa-mir expression")