practical broom
Yeast dataset
Aurelien Ginolhac, Eric Koncina
4 May 2017
This practical work is adapted from the exhaustive example published by David Robinson on his blog.
In 2008, Brauer et al. used microarrays to test the effect of starvation on the growth rate of yeast. For example, they tried limiting the yeast’s supply of glucose (sugar to metabolize into energy), of leucine (an essential amino acid), or of ammonium (a source of nitrogen).
Project - set-up
- Create a folder
data - Download
Brauer2008_DataSet1.tdsinside thedatafolder
Load the [Brauer2008_DataSet1.tds] file as a tibble named original_data. This is the exact data that was published with the paper (though for some reason the link on the journal’s page is broken). It thus serves as a good example of tidying a biological dataset “found in the wild”.
1 Tidying the data
Have a look at the dataset. Is the data “tidy”?
1.1 Many variables are stored in one column
- Gene name e.g. SFB2. Note that not all genes have a name.
- Biological process e.g. “proteolysis and peptidolysis”
- Molecular function e.g. “metalloendopeptidase activity”
- Systematic ID e.g. YNL049C. Unlike a gene name, every gene in this dataset has a systematic ID.
- Another ID number e.g.
1082129. We don’t know what this number means, and it’s not annotated in the paper. Oh, well.
- Use the appropriate function provided in the
tidyrlibrary to split these values and generate a column for each variable.
Tip
Special characters such as pipes (|) must be despecialized as they have a specific meaning. In R, you have to use 2 backslashes like \\| for one pipe |
- Once you separated the variables delimited by two “
||”, check closer the new values: You will see that they might start and/or end with whitespaces which might be inconvinient during the subsequent use.- To remove these whitespaces, R base provides a function called
trimws(). Let’s test how the function works: dplyrallows us to apply a function (in our casetrimws()) to all columns. In other words, we would like to modify the content of each column with the output of the functiontrimws(). How can you achieve this? Save the result in atibblecalledcleaned_data.
- To remove these whitespaces, R base provides a function called
# Creating test string with whitespaces:
s <- " Removing whitespaces at both ends "
s## [1] " Removing whitespaces at both ends "trimws(s)## [1] "Removing whitespaces at both ends"- We are not going to use every column of the dataframe. Remove the unnecessary columns:
number,GID,YORFandGWEIGHT.
Look at the column names.
Do you think that our dataset is now “tidy”?
1.2 Column headers are values, not variable names
- Keep care to build a tibble with each column representing a variable: At this point we are storing the sample name (will contain
G0.05…) as a different columnsampleassociated to values inexpressioncolumn. Save ascleaned_data_melt
Now look at the content of the sample column. We are again facing the problem that two variables are stored in a single column. The nutrient in the first character, then the growth rate.
Use the same function as before to split the sample column into two variables nutrient and rate (use the appropriate delimitation in sep.
Tip
Consider using the convert argument. It allows to convert strings to number when relevant like here.
1.3 Turn nutrient letters into more comprehensive words
Right now, the nutrients are designed by a single letter. It would be nice to have the full word instead. One could use a full mixture of if and else such as if_else(nutrient == "G", "Glucose", if_else(nutrient == "L", "Leucine", etc ...)) But, that would be cumbersome.
using the following correspondences and dplyr::recode, recode all nutrient names.
G = "Glucose", L = "Leucine", P = "Phosphate",
S = "Sulfate", N = "Ammonia", U = "Uracil"1.4 Cleaning up missing data
Two variables must be present for the further analysis:
- gene expression named as
expression - systematic id named as
systematic_name
delete observations that are missing any of the two mandatory variables. How many rows did you remove?
2 Representing the data
Tidying the data is a crucial step allowing easy handling and representing.
2.1 Plot the expression data of the LEU1 gene
Extract the data corresponding to the gene called LEU1 and draw a line for each nutrient showing the expression in function of the growth rate.
2.2 Plot the expression data of a biological process
For this, we don’t need to filter by single gene names as the raw data provides us some information on the biological process for each gene.
Extract all the genes in the leucine biosynthesis process and plot the expression in function of the growth rate for each nutrient.
2.3 Perform a linear regression in top of the plots
Let’s play with the graph a little more. These trends look vaguely linear.
Add a linear regression with the appropriate ggplot2 function and carrefully adjust the method argument.
2.4 Switch to another biological process
Once the dataset is tidy, it is very easy to switch to another biological process. Instead of the “leucine biosynthesis”, plot the data corresponding to sulfur metabolism.
Tip
you can combine the facet headers using + in facet_wrap(). Adding the systematic name allows to get a name when the gene name is missing.
3. Linear models
We can see that most genes follow a linear trend under a starvation stress. Instead of manipulating all data points, we would like to estimate just the 2 parameters of a linear model. What are they? And what do they represent for this experiment?
3.1 Perform all linear models
Perform a linear regression of expression explained by rate for all group of name, systematic_name and nutrient. By sure to convert the linear models to a tibble using broom as saved as cleaned_lm
The computation takes ~ 60 sec on my macbook pro. For test purposes, you may want to subsample per group a small number of genes
How many models did you perform?
bonus question: you have observed a warning
essentially perfect fit: summary may be unreliable, why? How can we clean up the dataset to avoid it?
3.2 Explore models, intercept values
For the gene LEU1, let’s look again at the linear models per nutrient and add as a dashed black line for mean of all intercepts.
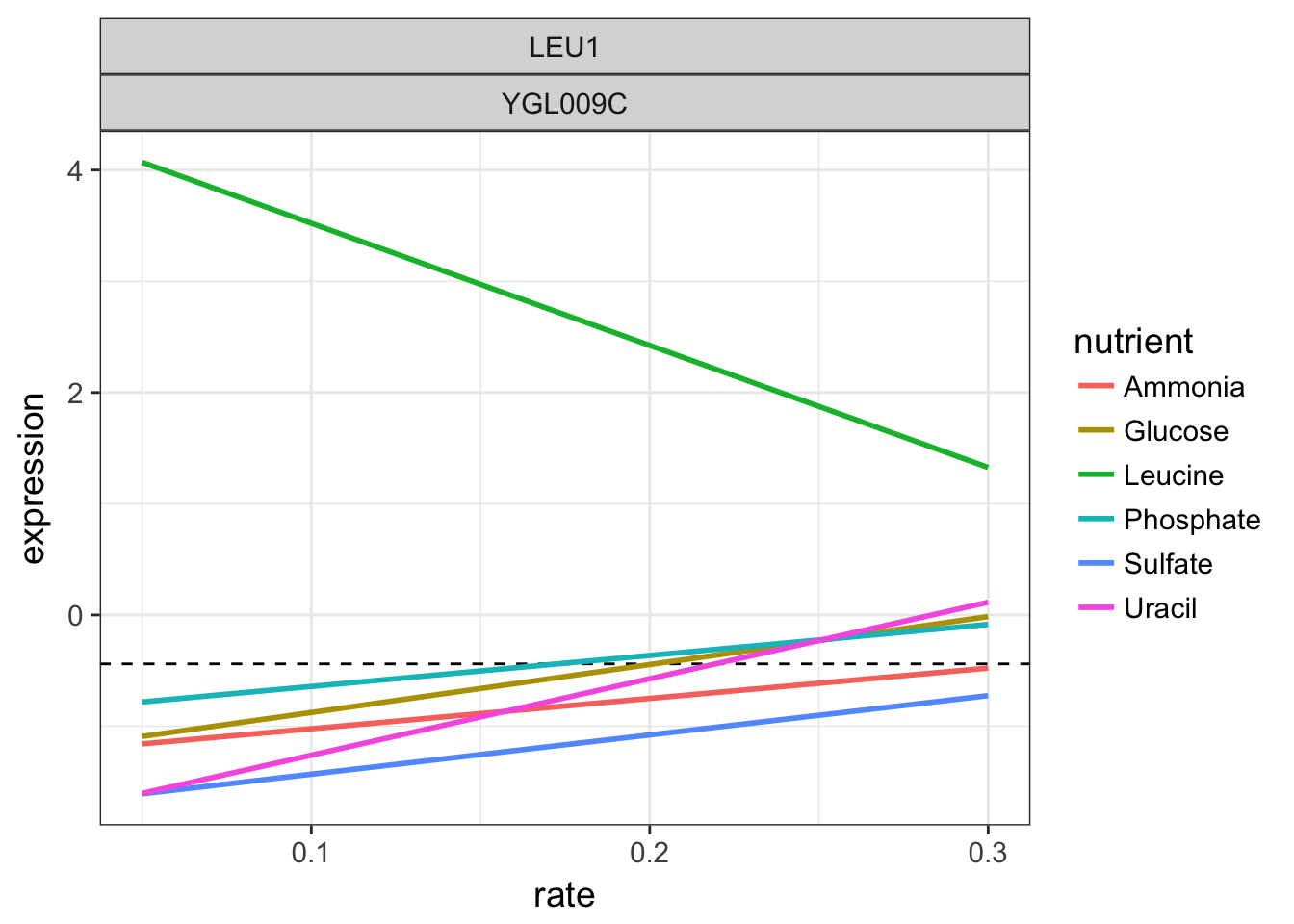
LEU1 linear models per nutrient
We can see that when leucine is the limiting factor, the yeast expresses massively this gene LEU1 to compensate. Remember that all experiments are performed at constant growth rates in this chemostat.
Now, compute the difference between the intercept and the mean(intercept) per systematic name, so for all nutrient per gene. And select the top 20 highest intercept values to their means. Save as top_20_intercept
- merge those 20 genes with the
cleaned_data_meltto retreived the original data points plot the data points and linear trends for those top 20 genes. Instead of the systematic name in the header, use the molecular function (
MF)what can conclude?
3.3 Explore models, slope estimates
We can now have a look at the slope estimates.
first, filter the term for slope estimate
rateand remove p-value that are missing. Save asrate_slopesdisplay the p-values histograms per nutrient
looking at those histograms, how do you estimate the ratio of null and alternative hypotheses?
how can retrieved the genes that belong most probably to the _alternative hypothesis?
display the histograms of both p and q values per nutrient